Medical device compliance software for every regulator
UDI COMPLIANCE & DATA MANAGEMENT SOLUTION
One source of truth for your medical device UDI data — submitted, synced, and provable across EUDAMED, GUDID, Swissdamed, SFDA, AusUDI and beyond.
Every regulatory base — your perimeter, your pace.
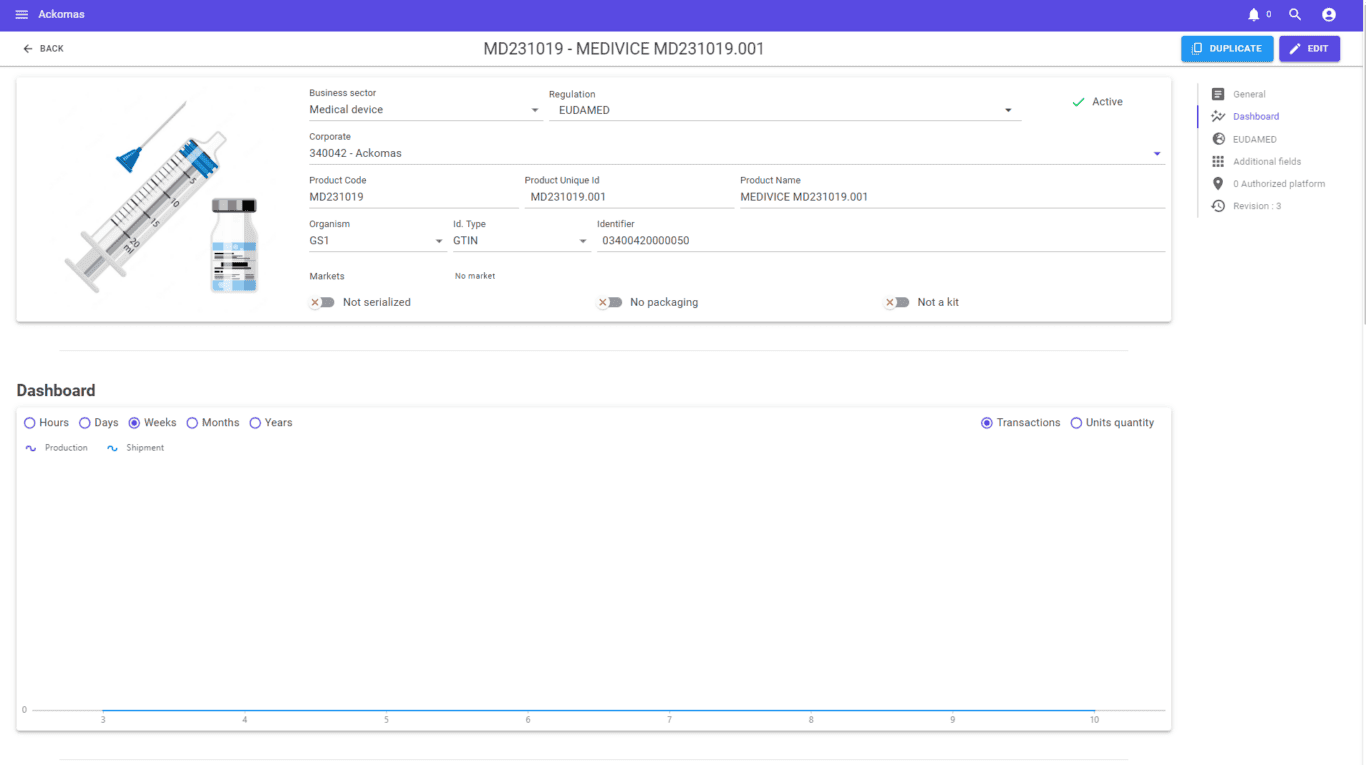
Trusted by medical device manufacturers — from single-base EU operators to multi-base international groups










Built for all medtech
Single-base teams to international groups
Regulatory truth, not data noise
Clean, governed, auditable
RA-led platform & support
Built and run by regulatory experts
Every regulator, one workspace
EUDAMED, GUDID, Swissdamed, SFDA, AusUDI and beyond
Regulatory data is fragmented. Each database asks for different data — in its own format.
Whether you manage one regulatory base or five, the day-to-day looks the same: device data kept by hand, in spreadsheets, re-keyed for every portal. Add a regulator and the work doesn't add up — it multiplies.
Multiple databases, same data, wasted effort
Teams rebuild the same master dataset from scratch every time a new regulator enters their map.
Inspections now check data quality
Since February 2026, FDA Program 7382.850 treats GUDID data quality as an audit criterion. EUDAMED Notified Body audits do the same.
No single source of truth, no audit-readiness
When data lives in spreadsheets and parallel platforms, no one can prove what was published, when, and by whom.
One workspace. Every regulator. Audit-ready by design.
WHAT ACKOMAS DOES
For every problem above, an answer. Govern your data once, keep it audit-ready, and publish to each regulator without re-keying.
Your device data
One master catalog — entered once, owned by your team.
One master catalog, every base
Regulatory evidence & traceability
Automated publishing to every regulator
Three plans. Every scale. One source of truth.
ACKOMAS is available in three plans. The difference is not the number of devices you manage — it is how complex your regulatory organization is. Pricing is shared after a 30-minute scoping call.
Essential
Single-base EU operations
EUDAMED only · up to 800 UDIs · up to 2 SRNs · instant compliance checks
Professional
Multi-country operations
EUDAMED + a 2nd regulation · up to 5,000 UDIs · up to 3 SRNs · advanced automation
Enterprise
International groups
Multi-regulatory environment · unlimited UDIs & SRNs · audit-trail, validation workflow & API access
RESOURCES
Built by regulatory experts who write what they know
Articles, guides, and webinars from the team building ACKOMAS. Sharp regulatory analysis, step-by-step playbooks, and live deep-dives — for RA/QA teams who want signal, not noise.
EUDAMED Registration: the complete step-by-step guide (2026)
Complete playbook to register medical devices in EUDAMED: UDI-DI, Basic UDI-DI submission, deadlines, legacy devices.
GUDID at the heart of the FDA framework
FDA's Compliance Program 7382.850 makes GUDID an inspectable, mandatory requirement.
How Santex Spa secured MDR compliance with ACKOMAS
Exclusive client reveal: MDR & EUDAMED compliance journey.
Let’s see how ACKOMAS can fit and scale with your scope
Tell us about your perimeter through the form. Our team gets back to you within 48h with the right person and the right next step.
The regulatory point
Sharp regulatory analysis across every regulator — why compliance is an ongoing discipline, not a one-off, and how to stay audit-ready as your data evolves.
Written by our regulatory team · Twice a month · Unsubscribe anytime